Synthetic quickstart
Above: the dataset produced by this example, scrolling through its spectral channels in the GUI.
This example generates a small, fully reproducible hyperspectral TIFF stack you can use to test installation and learn the full GUI workflow — without any experimental data. It is the canonical "does everything work?" check shipped with HS-MOSAIC.
Use it when:
- you want to confirm that TIFF loading, spectral-axis detection, ROIs, seeds, NNMF/NNLS, and export all work end-to-end,
- you want a safe dataset for learning the workflow before touching real data,
- you need a repeatable dataset for screenshots, GIFs, or bug reports.
TL;DR
# 1. generate the data
python docs/examples/generate_synthetic_quickstart.py --output synthetic_quickstart_data
# 2. launch the GUI
hs-mosaic # or: python -m hs_mosaic
# 3. in the GUI: load synthetic_hs_stack.tif → Load Spectrum from File on the CSV
# → set Components = 5 → NNMF + Custom init + Fixed-H NNLS mode
# → Run Analysis
The rest of this page explains what the dataset contains, what to expect at each step, and the seed experiments worth trying.
Synthetic data generation
Run the generator script from an environment where the application dependencies are installed:
python docs/examples/generate_synthetic_quickstart.py --output synthetic_quickstart_data
On Windows, if python is not available but the Python launcher is installed:
py docs/examples/generate_synthetic_quickstart.py --output synthetic_quickstart_data
The output folder contains:
| File | Purpose |
|---|---|
synthetic_hs_stack.tif |
3D hyperspectral stack with shape (channel, y, x). |
wavelength.json |
Spectral-axis metadata loaded automatically by the GUI. |
synthetic_reference_spectra.csv |
One reference spectrum per synthetic component, importable with Load Spectrum from File. |
README.txt |
Short description of the generated data. |
The default command creates a microbead-like mixture with five spectral components (lipid-like, two mutated lipid-like variants, protein-like, and a broad background). You can make the field denser or add more mutated lipid-like variants (which raises the component count):
python docs/examples/generate_synthetic_quickstart.py --output synthetic_quickstart_data --beads-per-class 14 --mutant-variants 5 --mutant-beads-per-variant 6
Useful generator options:
| Option | Default | Effect |
|---|---|---|
--beads-per-class |
12 |
Number of ordinary lipid-like and protein-like beads. |
--mutant-variants |
2 |
Number of mutated lipid-like spectral variants. Each variant shares the lipid peak but has a different tail. Raising this raises the total component count. |
--mutant-beads-per-variant |
4 |
Number of beads for each mutated lipid-like variant. |
--seed |
7 |
Random seed for bead positions and noise. |
--noise |
280 |
Gaussian noise level added to the stack. |
What the dataset contains
The default stack has five synthetic components (lipid-like, two mutated lipid-like variants, protein-like, and a broad background):
| Component | Spatial pattern | Spectrum |
|---|---|---|
| Lipid-like | Several bead-like spots | Narrow peak near 2850 cm⁻¹ |
| Mutated lipid-like variants | Several smaller bead-like spots per variant | Same main 2850 cm⁻¹ peak as lipid-like, but variant-specific high-wavenumber tails |
| Protein-like | Several bead-like spots | Main peak near 2930 cm⁻¹ plus a weaker overlapping peak near 2850 cm⁻¹ |
| Broad background | Smooth gradient over the field | Broad low-frequency spectral background |
The data are intentionally simple but not perfectly separated. The lipid-like, mutated lipid-like, and protein-like spectra all contribute around 2850 cm⁻¹, so inspecting only that channel is ambiguous. The mutated lipid-like beads are deliberately artificial cases: they share the lipid peak but have different tails. This makes the dataset look more like a microbead mixture and demonstrates why full-spectrum NNMF/NNLS can distinguish signals that look similar in one channel.
A good multivariate analysis should group ordinary lipid-like beads, separate the mutated lipid-like bead variants, recover protein-like beads, and keep the smooth background as its own component.
Why seeded NNMF or fixed-H NNLS is useful here
This dataset is designed so that a single bright channel is not enough. Around 2850 cm⁻¹, several components light up at the same time:
- ordinary lipid-like beads,
- mutated lipid-like beads,
- part of the protein-like signal.
The separation comes from the full spectral shape. The mutated lipid-like variants have the same main lipid peak, but different tails. The protein-like signal partly overlaps with lipid at 2850 cm⁻¹, but its strongest information is closer to 2930 cm⁻¹. Seeded NNMF and fixed-H NNLS use these full-spectrum differences instead of treating one channel as one component.
Use Fixed-H NNLS mode first if you want a reference result: load the provided spectra and keep them fixed. This shows what the GUI can recover when the spectra are already known.
Then experiment with seeded NNMF by disabling Fixed-H NNLS mode while keeping NNMF and Custom initialization enabled. In this mode, seeds are starting points rather than fixed rules. The result can adapt, which is useful when real reference spectra are approximate or when the sample spectrum differs from the library spectrum.
Three ways to analyze this data (illustrated)
The clips below walk the same dataset through three approaches, from a deliberately wrong first attempt to two clean separations. They are the fastest way to build intuition for how component count and seeding drive the result.
1. Too few components: the species mix
Start with plain random NNMF (no seeds) but set the component count too low: 3, when the data actually contains five components (four bead species plus a background). With too few components to go around, the solver is forced to pack several species into one component and mix others (see purplish blops).
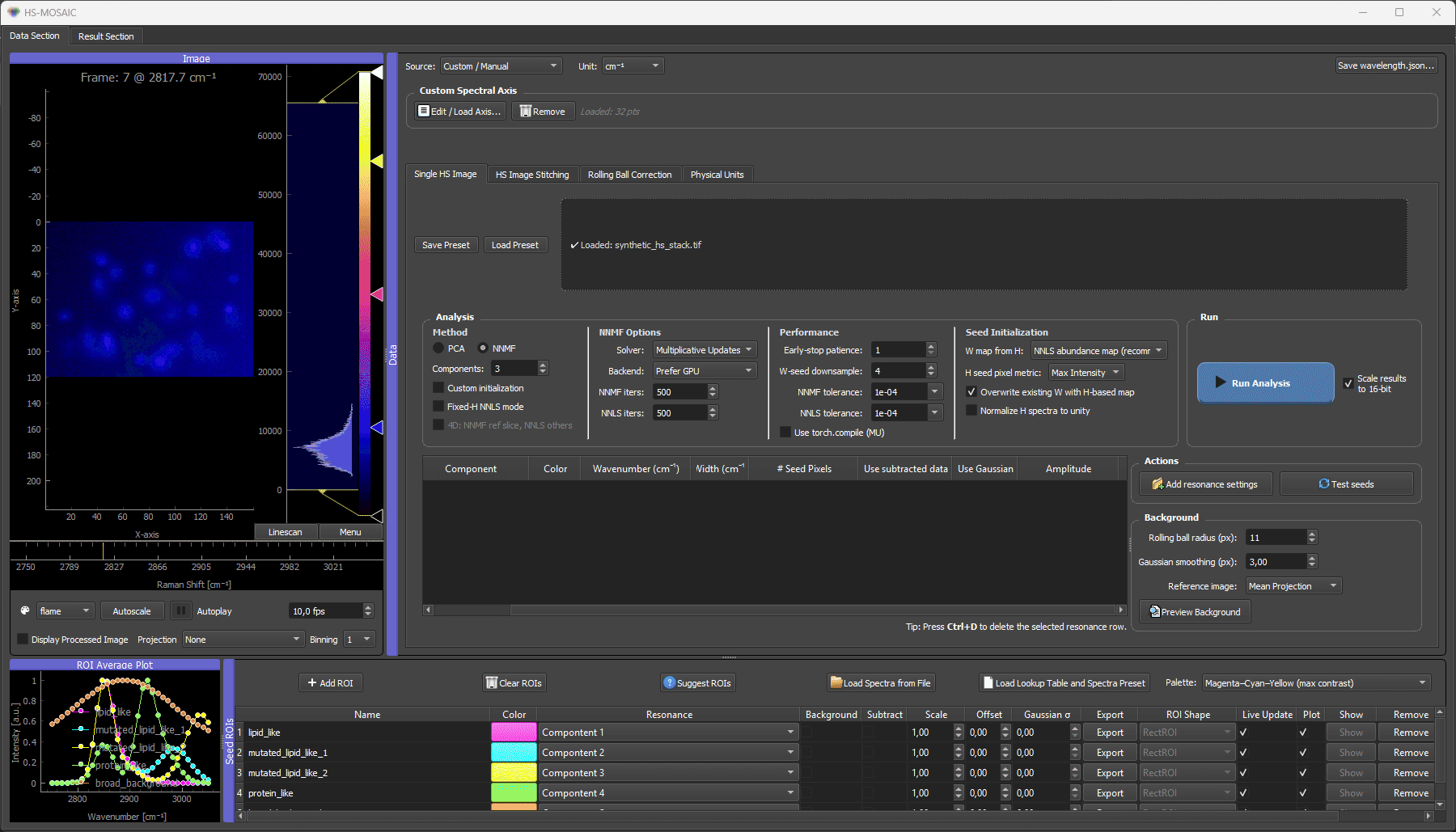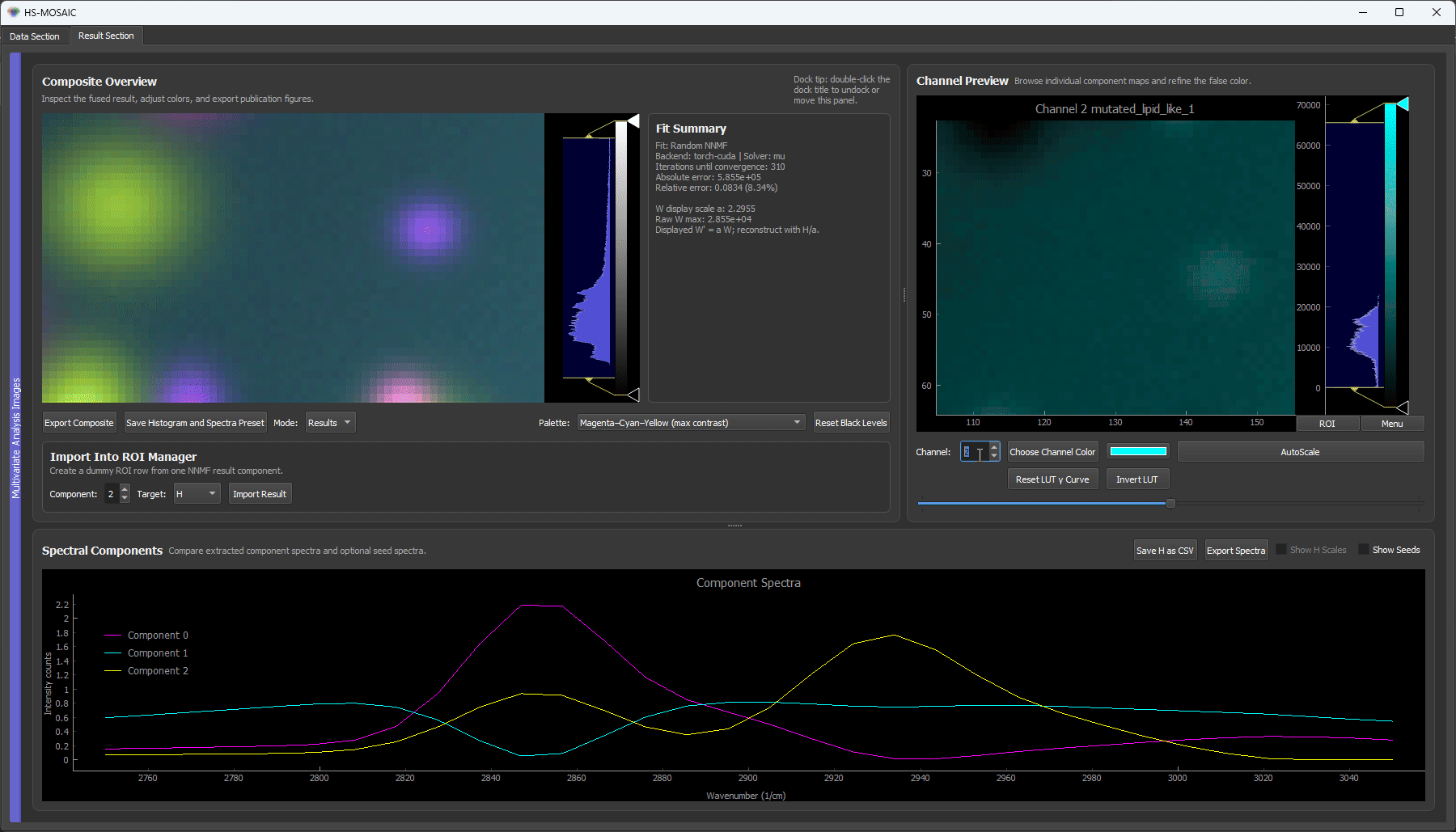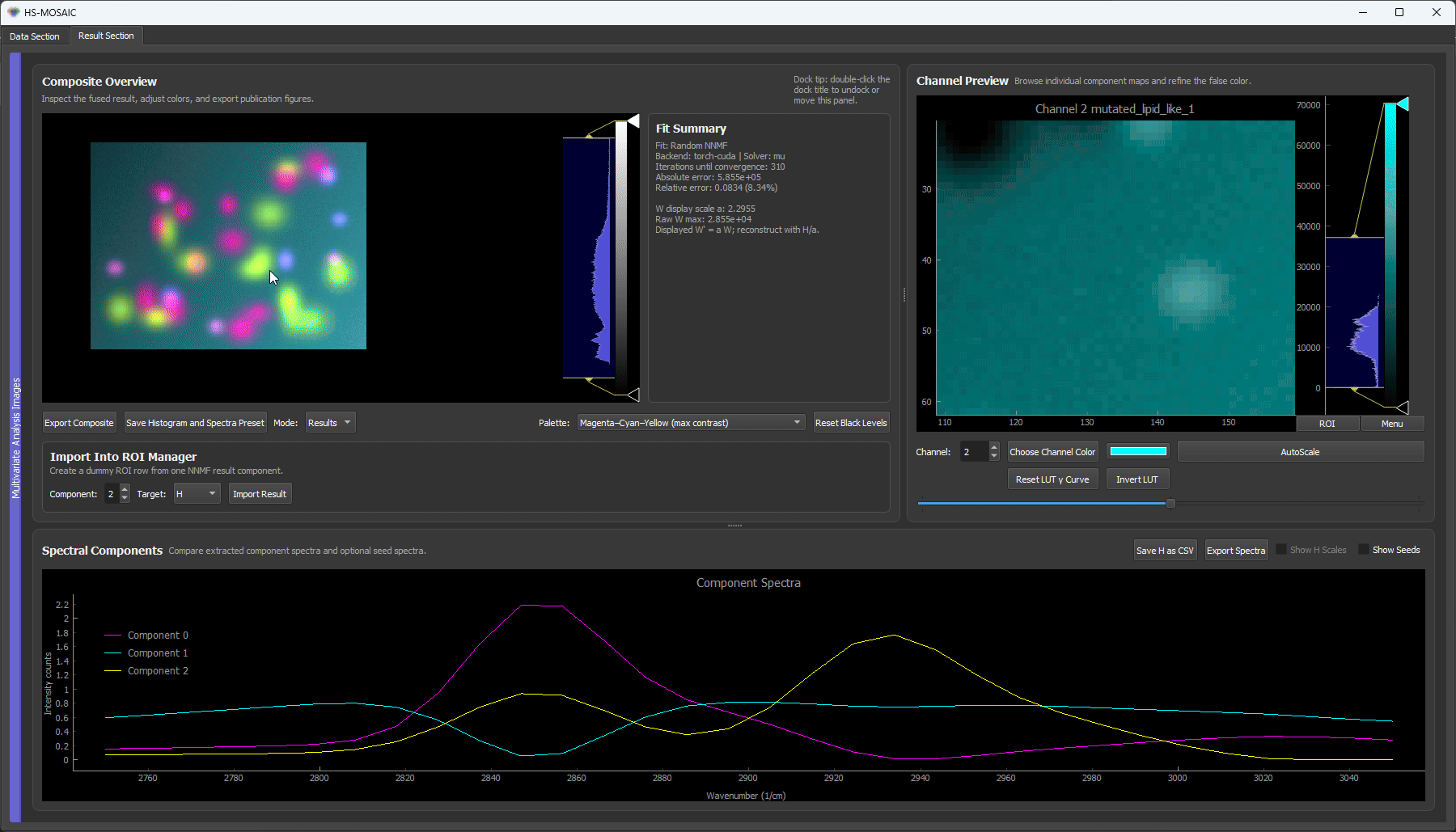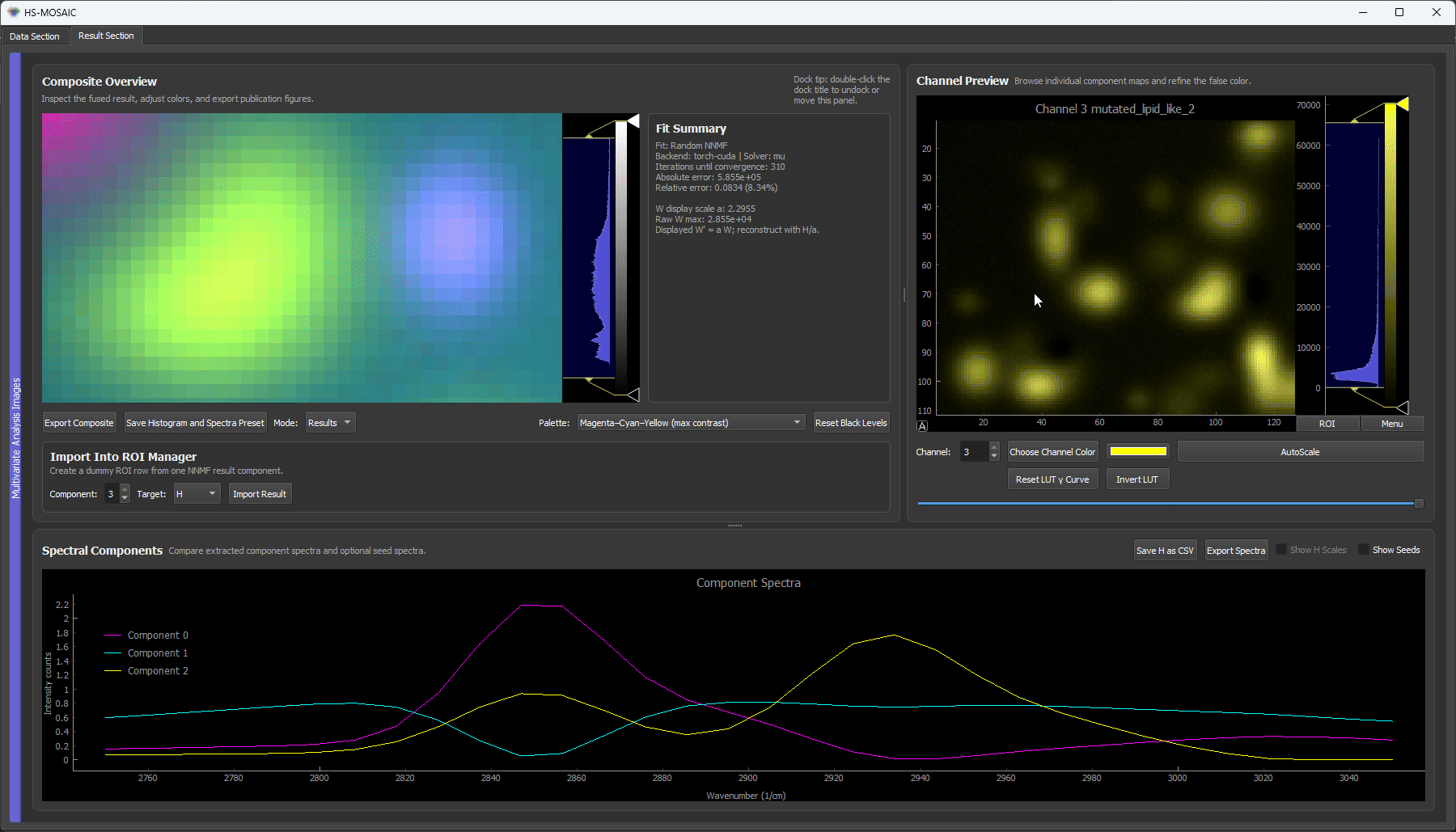
Random NNMF with 3 components. There are not enough components to hold each species separately, so the yellow and cyan beads are merged into a shared component, and the smooth background bleeds heavily into the cyan component. No amount of seeding fixes this while the count is below the true number of components. The takeaway: when species are mixed or the background is unaccounted for, the first thing to check is whether the component count is high enough.
2. The full guided workflow: first guess, then seed, then refine
Now use what the failed run taught us and set the count to 5. This clip runs the complete guided workflow end to end:
- Random NNMF for a first guess with the correct component count (5).
- Turn on the Composite (from analysis) projection mirror in the raw image viewer, so the live false-color composite sits next to the raw data for inspection.
- Draw ROIs on the bead species, using the random result as a guide for where each species sits. One species (a dark bead) is not cleanly recovered by the random init, so it is found directly on the image and given its own ROI.
- Run custom-initialized NNMF with the four bead ROIs seeded. The fifth component, the smooth background, is filled automatically from the data residual (no ROI needed for it).
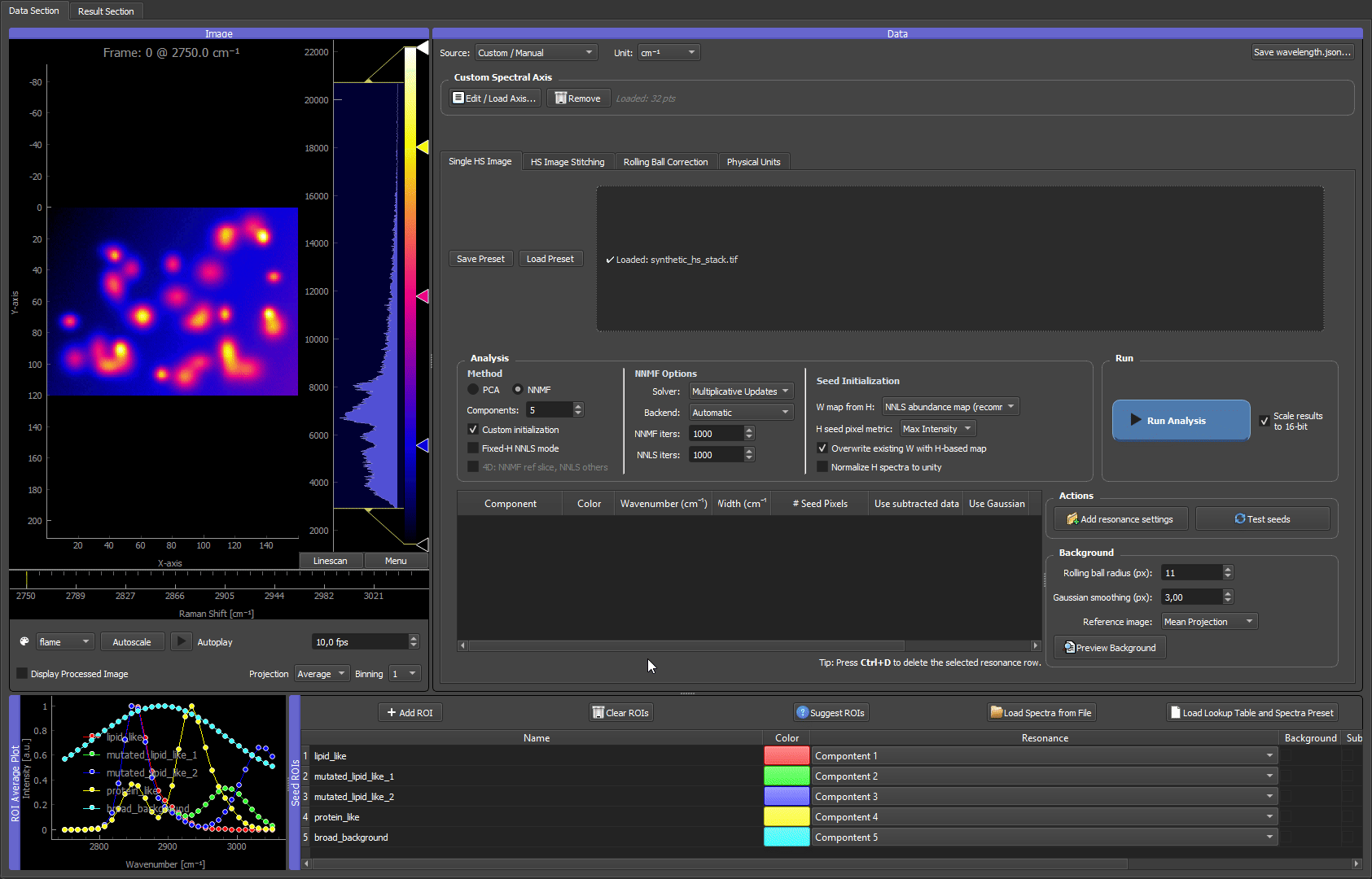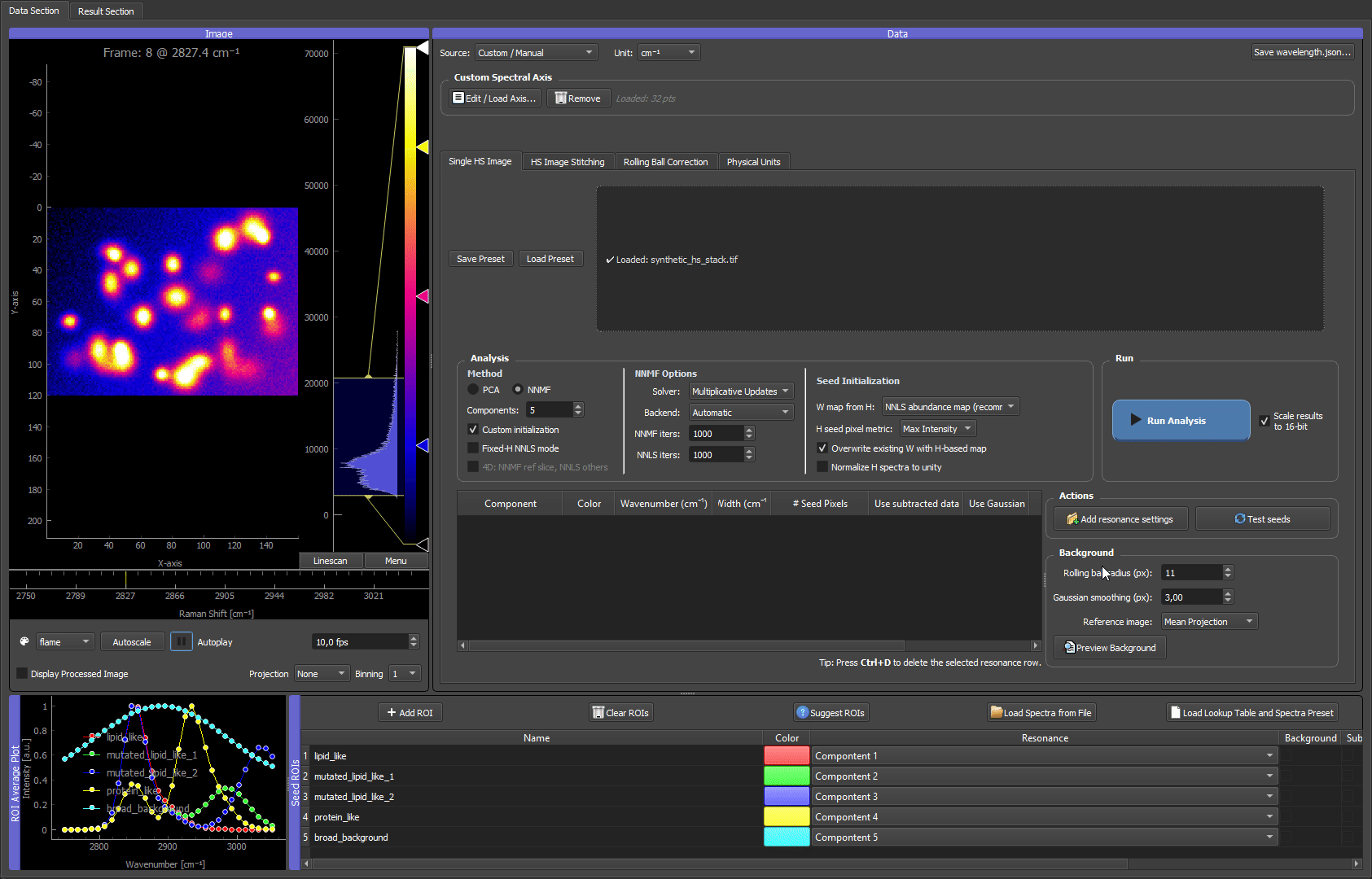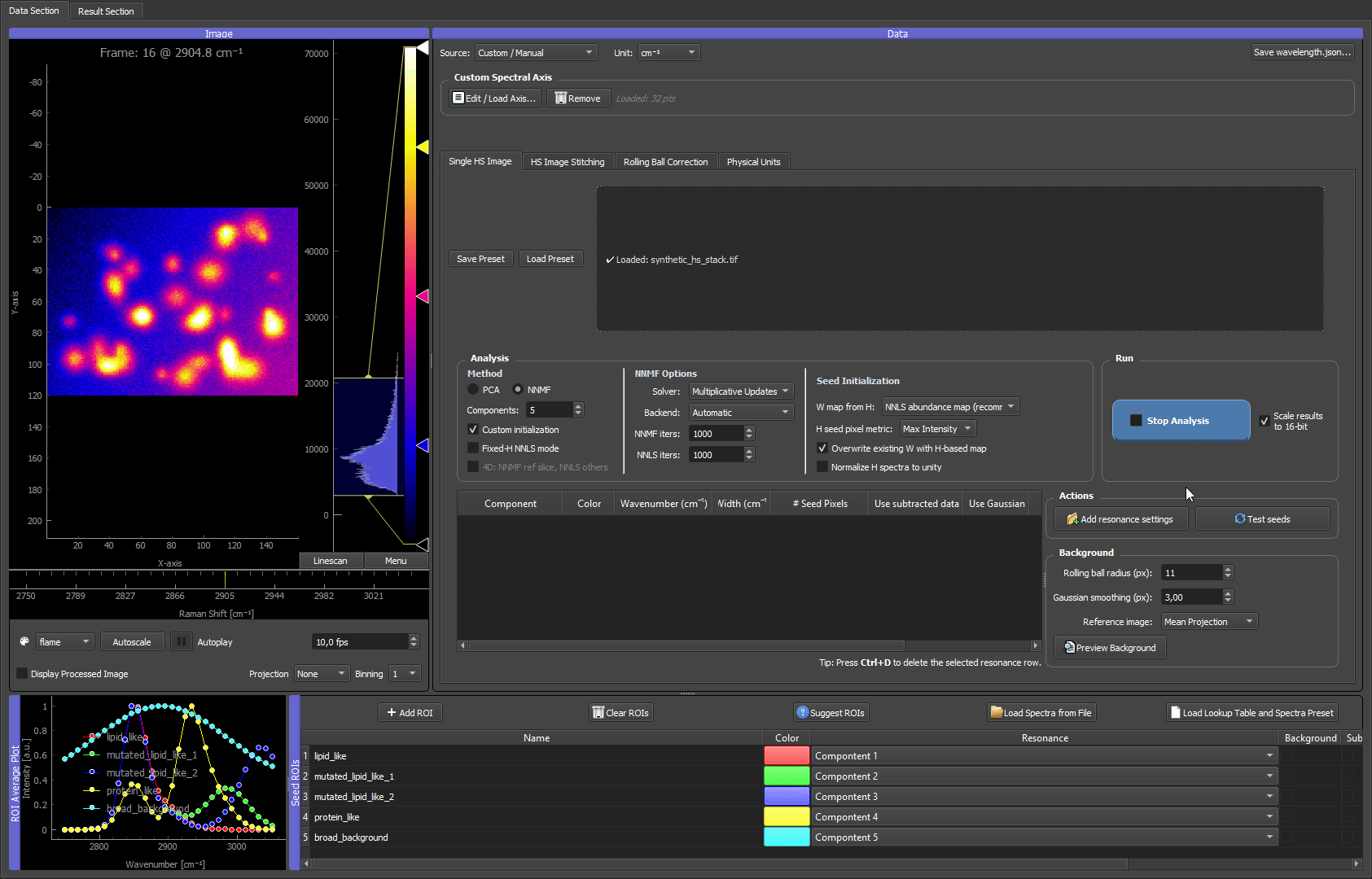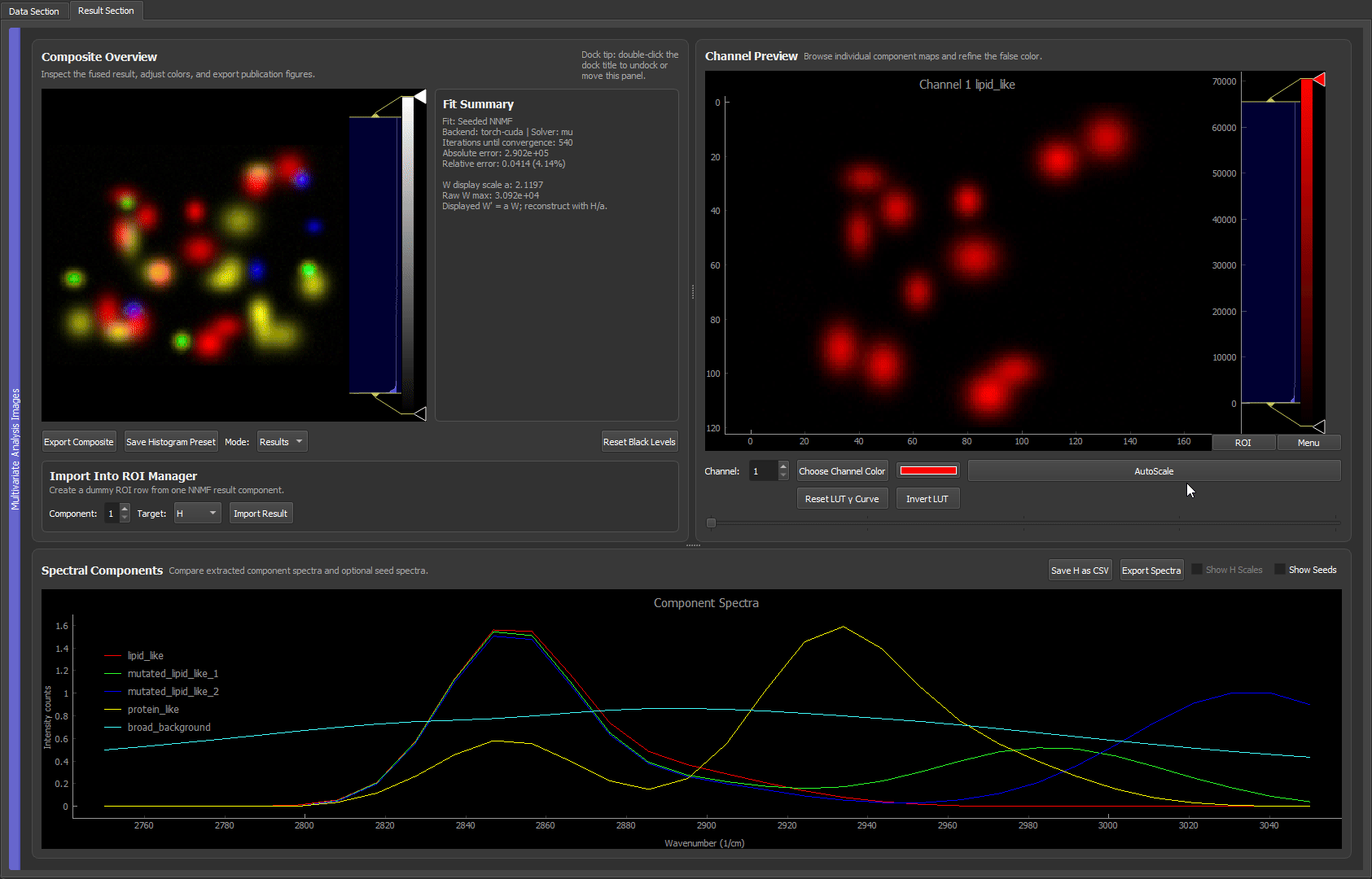
- Result: every bead species lands in its own dedicated false-color channel, and the background is cleanly separated into the last component.
- Bonus: set the background spectrum in the final optimization step
The full loop: a random first guess shows roughly what is in the data, ROIs lock in the four bead species (including the one the random run missed), and a final custom NNMF separates all five components cleanly, with the background recovered from the residual.
3. Reference spectra: the spectrometer case
When you already have trustworthy reference spectra (measured on a spectrometer, taken from a library, or exported from a prior experiment), you can skip the ROI hunting entirely. Load synthetic_reference_spectra.csv with Load Spectrum from File, assign each loaded spectrum to its component, and run custom-initialized NNMF.
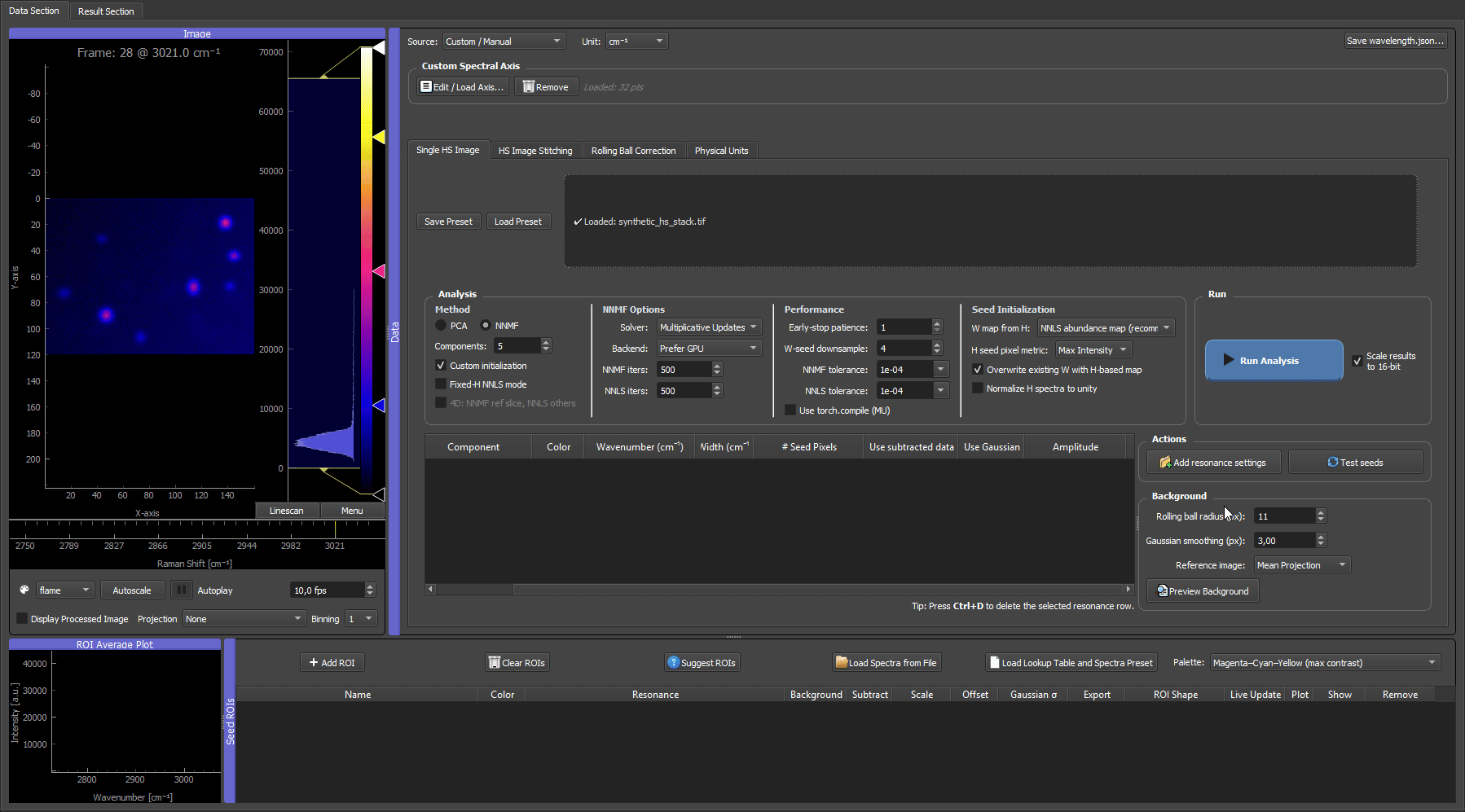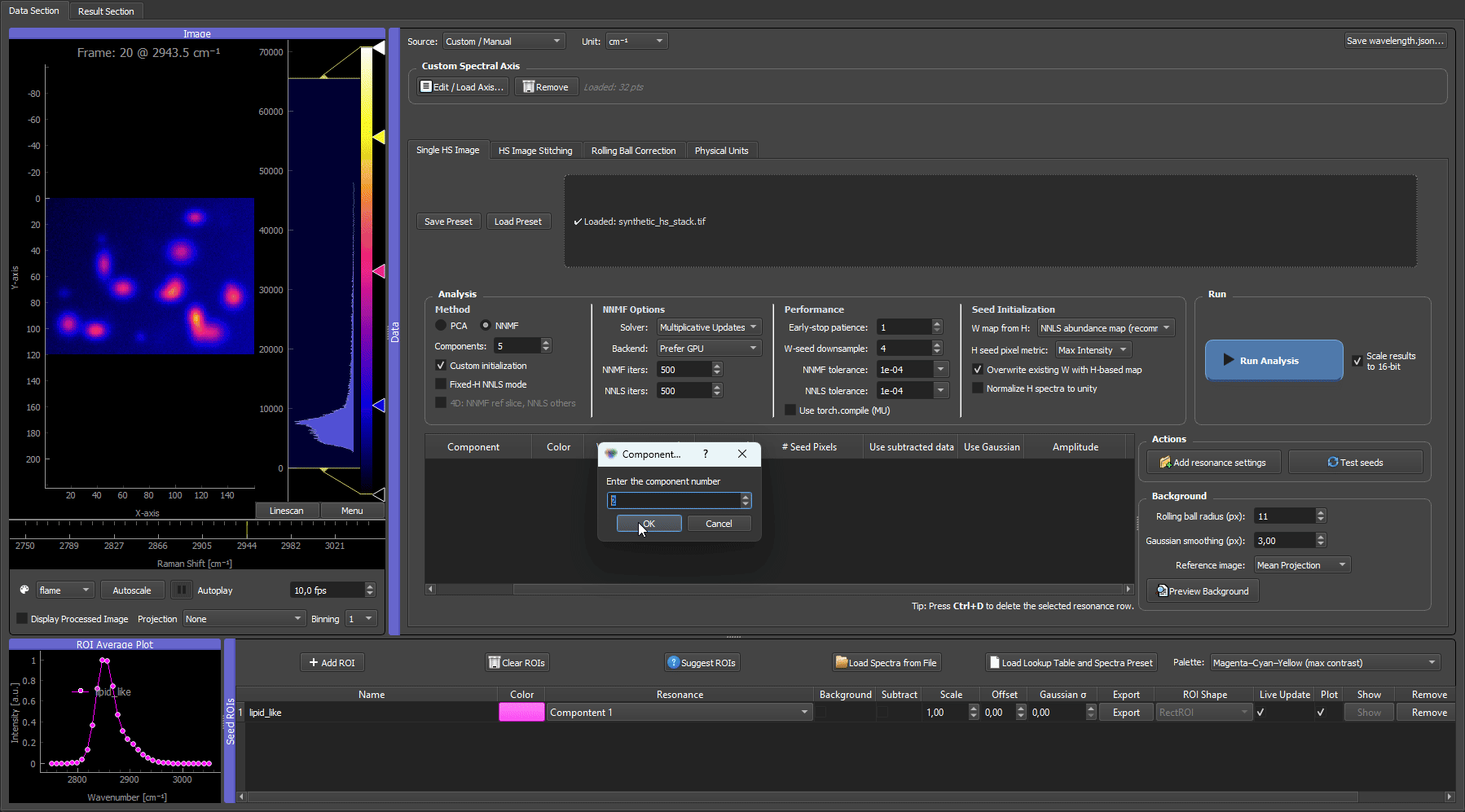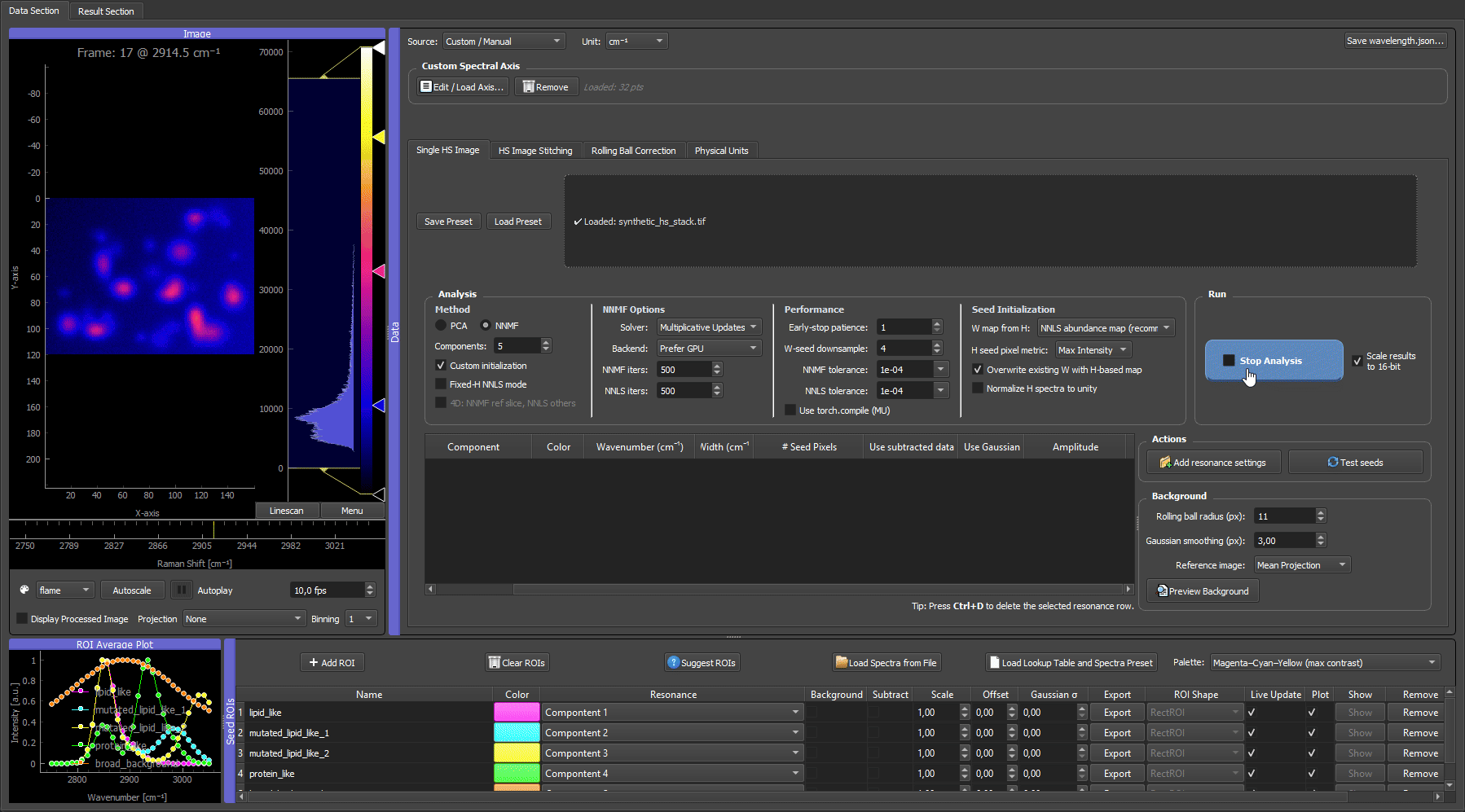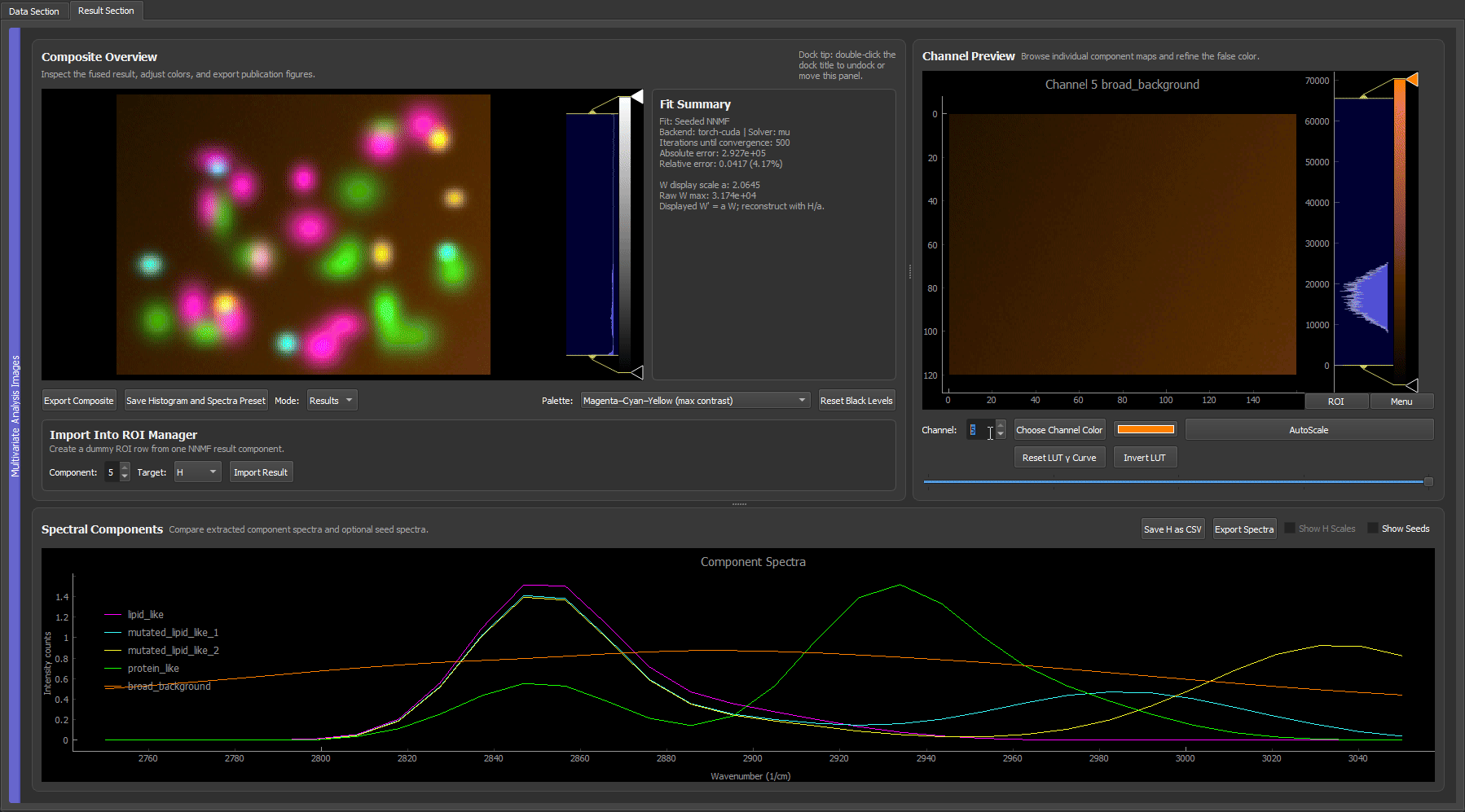
Loading the reference spectra, assigning them to components, and running custom NNMF. Because the spectra are already trusted, the separation is clean immediately, with each bead species in its own false-color channel and no iterative ROI work needed. This mirrors a real experiment where the pure-component spectra are known up front.
Both the ROI-seeded run (clip 2) and the reference-spectra run (clip 3) reach the same goal: great separation, with each bead kind in its own dedicated false-color channel.
Try the different W-seed modes — NNLS is the most specific
When you seed only spectra (clips 2 and 3), the GUI builds each component's starting spatial map from its spectrum, and the W map from H dropdown in the Seed Initialization controls decides how. It is worth switching between the modes on this dataset to see the effect:
- NNLS abundance map (the default, recommended) is the most specific mode. It solves a full non-negative least-squares fit against all seeded spectra at once, so every pixel is explained by as few components as possible and the maps come out clean and near-binary. This is exactly why the beads in clips 2 and 3 land so crisply in their own channels: the bead species occupy different pixels, which is the regime where NNLS shines.
- Selective score is softer. It favors the target spectrum but does not force one winner per pixel, so it is the better choice when components genuinely share pixels (co-localized signals, fluorophore mixtures inside one voxel).
- H-weighted average, Average image, and Homogeneous (empty) are neutral or legacy fallbacks, useful mainly when NNLS is unstable or when you want a component discovered from scratch.
For these well-separated beads, NNLS is the right default and the most discriminating choice. On data where species overlap within the same pixels, compare NNLS against selective score and pick whichever gives the more plausible maps. See Picking nnls vs selective_score.
Why resonance/spectral-info seeding is not a good fit for this dataset
Seeding from the spectral-information table (a resonance center plus width, used for Gaussian seeds and seed pixels) works best when each component owns a distinct peak position. That is not the case here: the lipid, mutated-lipid, and part of the protein signal all contribute around 2850 cm⁻¹, and the lipid and mutated-lipid beads share their dominant peak almost entirely. A single resonance position cannot isolate one species, because the species are told apart by their full spectral shape, not by a unique peak. For this kind of strongly overlapping data, prefer ROI seeds (clip 2) or loaded reference spectra (clip 3). Gaussian/seed-pixel resonance seeding is better suited to datasets where each component has a clearly separated band (see Seed pixels and Gaussian seeds in Essentials).
Seed experiments to try
| Seed strategy | What to do | What it teaches |
|---|---|---|
| Reference spectra | Load synthetic_reference_spectra.csv with Load Spectrum from File. |
Best controlled starting point; useful for checking the expected separation. |
| Spatial ROIs | Draw ROIs on representative lipid-like beads, mutated lipid-like beads, protein-like beads, and a background region. | Shows how well ROI-derived mean spectra work when you choose representative regions. |
| Gaussian models or seed pixels | Add resonance settings near 2850 cm⁻¹, 2930 cm⁻¹, and the mutated lipid tail region around 2985-3035 cm⁻¹. | Shows how approximate peak knowledge can guide analysis without external spectra. Note: this is the weakest fit for this dataset, because lipid and mutated-lipid share their dominant 2850 cm⁻¹ peak; a single resonance position cannot separate them (see the note above). Prefer ROI seeds or loaded reference spectra here. |
| Mixed strategy | Use loaded spectra for known components, then draw or model only the uncertain component. | Mirrors real workflows where some components are known and others need exploration. |
| No seeds / random NNMF | Disable Custom initialization and run NNMF. | Shows why unguided NNMF can be less stable when components overlap. |
The goal is not only to get one correct answer. Use this dataset to see how seed quality, component count, and fixed-vs-adaptive spectra change the result.
GUI workflow
- Start the GUI.
- In the Single HS Image tab, load
synthetic_hs_stack.tif. - Confirm that the spectral axis is loaded from
wavelength.json. - Set Components to the number of reference spectra in the CSV. With the default generator settings, use
5. - Click Load Spectrum from File in the ROI Manager and select
synthetic_reference_spectra.csv. - Assign the loaded spectra to the matching component numbers if prompted.
- In the Analysis panel, select NNMF, keep Custom initialization enabled, and enable Fixed-H NNLS mode for the first reproducible test.
- Click Run Analysis.
- Inspect the result viewer:
- lipid-like components should map bead-like spots with the ordinary lipid spectrum,
- mutated lipid-like components should map smaller bead-like spots with different tails,
- the protein-like component should map beads with stronger 2930 cm⁻¹ signal,
- the background component should look like a smooth gradient.
For a more exploratory test, disable Fixed-H NNLS mode and run seeded NNMF with the same spectra. The H spectra may adapt slightly because seeded NNMF treats seeds as starting points rather than fixed spectra.
To demonstrate the point of the synthetic data, compare the raw channel around 2850 cm⁻¹ with the NNMF/NNLS component maps. The raw channel lights up many beads at once, while the multivariate result separates ordinary lipid-like beads, mutated lipid-like variants, and protein-like beads by using the whole spectral shape.
Expected outcome
| Check | Expected result |
|---|---|
| TIFF loading | First channel appears in the raw image viewer. |
| Spectral axis | The x-axis uses Raman-shift values from wavelength.json. |
| Spectrum import | One dummy ROI row appears for each reference spectrum in the CSV. |
| Fixed-H NNLS | H spectra stay equal to the imported references. |
| Seeded NNMF | Maps remain similar, but H spectra may adapt. |
| Overlap demonstration | The 2850 cm⁻¹ channel is ambiguous, but the separated component maps distinguish ordinary lipid-like beads, mutated lipid-like variants, and protein-like beads. |
| Export | Save H as CSV and Export Composite should both produce usable files. |
What to look at if something looks wrong
| Symptom | Likely cause | Fix |
|---|---|---|
| Image opens but spectral axis is in channel indices, not cm⁻¹ | wavelength.json was not placed next to the TIFF, or was renamed. |
Re-run the generator into a folder that already contains the TIFF, or copy the JSON next to it manually. See Spectral axis reference. |
| "Loaded image contains NaN or Inf" warning | Should never appear for the generated stack. If it does, the TIFF was rewritten by another tool. | Re-generate from scratch. |
| Fewer or more bead components than expected | Component count in the Analysis panel does not match the CSV. | Set Components to the number of spectra columns in synthetic_reference_spectra.csv (default 5). |
| Composite map looks all-cyan or all-grey | A background or fixed-W ROI dominates the display, or all components share the same colour. | Open the ROI manager, give each component a distinct LUT colour, and hide the background component for visual checks. |
| Fixed-H NNLS maps look much grainier than seeded NNMF | This is expected behaviour, not a bug. | See the explanation in Analysis modes – Fixed-H NNLS. |